Molecular traffic jam may underlie rare kidney disease, other protein misfolding disorders
Researchers find that several genetic disorders, including MUC1 kidney disease, may share a novel molecular mechanism — and identify a promising therapeutic lead
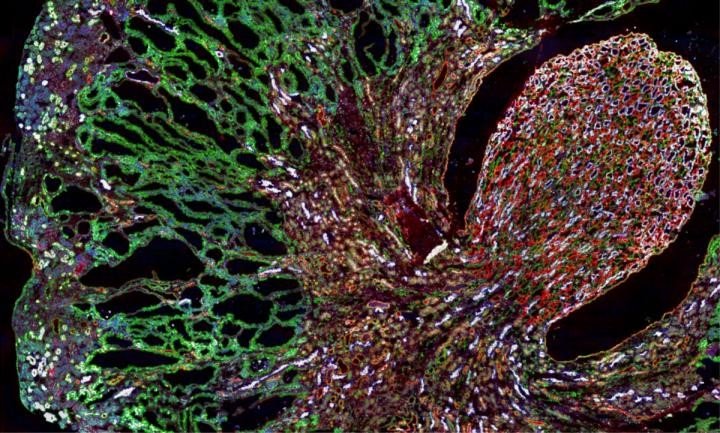
Misfolded MUC1-fs protein (green) builds up in the kidneys of a mouse model of MUC1 kidney disease.
Researchers have long known that dozens of inherited diseases, called toxic proteinopathies, are caused by the build-up of specific misfolded proteins in cells. But, the molecular mechanisms responsible for this accumulation have remained a mystery — hampering efforts to develop therapies.
Now, researchers at the Broad Institute of MIT and Harvard and Brigham and Women’s Hospital (BWH), writing in the journal Cell, have discovered that some of these toxic proteinopathies may arise from a single, previously unrecognized cause: a jam at a specific step in a cellular shipping network called the secretory pathway, which delivers proteins either to the cell surface or one of the cell’s protein-disposal systems.
The discovery came in a study of a rare disorder called MUC1 kidney disease (MKD), which the researchers showed stems from the harmful build-up of a misfolded version of a protein called MUC1 in kidney cells. Moreover, the researchers found that the cell’s failure to remove the misfolded protein could be traced to a specific molecular step in the secretory pathway — and, remarkably, that a compound called BRD4780 could clear the traffic jam. The compound worked in human kidney cells and kidney organoids (miniature kidneys-in-a-dish engineered from patient cells), and in an animal model of MKD. BRD4780 could be a starting point for developing new medicines for MKD and several other toxic proteinopathies, for which no treatments are currently available.
“What blew my mind is that, in the process of studying the mechanism behind MKD, we have uncovered fascinating new biology about how cells handle misfolded proteins, and these key insights may help us address several devastating diseases,” said study senior author Anna Greka, an institute member and director of the Broad’s Kidney Disease Initiative (KDI), an associate professor at Harvard Medical School, and a kidney specialist at BWH. “Our team is working around the clock to translate these discoveries into a new therapy we can bring to patients as quickly as possible.”
TRANSPORTATION BREAKDOWN
MKD is a rare, inherited disease that leads to kidney failure. In 2013, a team led by Broad president and founding director Eric Lander and institute member and Program in Medical and Population Genetics co-director Mark Daly tracked down the genetic root of MKD (previously called medullary cystic kidney disease, or MCKD): the addition of a single letter in the gene mucin 1 (MUC1).
The mutation results in the production of a truncated, misfolded protein dubbed MUC1-fs that collects in patients’ kidney cells. As MUC1-fs accumulates, kidney cells die, eventually leading to kidney failure.
To find drug-like compounds that might help clear MUC1-fs from cells, first author and postdoctoral fellow Moran Dvela-Levitt of the KDI, Greka and their colleagues turned to the Broad Drug Repurposing Hub — a collection of more than 3,700 compounds at different stages of the drug development process, maintained by the institute’s Center for the Development of Therapeutics. Their search turned up BRD4780, a compound that never made it to the clinic, cast aside when it proved to be unsuccessful as a blood pressure drug. Greka, Dvela-Levitt, and their colleagues found that BRD4780 eliminated MUC1-fs, left normal MUC1 untouched, and prevented kidney cells with the MKD mutation from dying.
When the researchers looked closely, they were surprised to find that BRD4780 does not bind to MUC1-fs. Rather, it latches onto TMED9, a so-called ‘cargo receptor’ that carries protein cargo along the secretory pathway.
Experiments in human kidney cells, an MKD mouse model, and patient-derived kidney organoids showed that the cargo receptor traps MUC1-fs. This keeps the misfolded protein from reaching the lysosome (an organelle whose job is to chew up unwanted proteins), causing MUC1-fs to rise to dangerous levels in kidney cells.
The researchers noted that when BRD4780 bound to TMED9, the cargo receptor released its hold on MUC1-fs, allowing the cell to degrade the misfolded protein. Knocking out the gene for TMED9 using CRISPR had the same effect.
“This is completely new biology,” Greka said. “We did not know that a cargo receptor like TMED9 could block and ultimately interfere with the destruction of a misfolded protein. And the question became, is the same biology at work in other conditions caused by a build-up of misfolded proteins?”
BEYOND THE KIDNEY
More than 50 diseases are considered toxic proteinopathies, including retinitis pigmentosa (RP, an inherited form of blindness where the retina degenerates) and UMOD-associated kidney disease (UKD, another rare genetic kidney disorder). Greka and her colleagues reasoned that similar problems with the secretory pathway might be to blame in at least a few of these diseases.
In in vitro experiments, the researchers saw that BRD4780 could reduce misfolded protein levels and increase cell survival in RP and UKD cells. Greka and her colleagues estimate that drugs similar to BRD4780 might reverse roughly 20 diseases where misfolded proteins become trapped early in the secretory pathway.
“Many of these disorders may tie back to the same mechanism,” Greka said. “Our next step is to develop a deeper understanding of cargo receptors, why they stop misfolded proteins from being eliminated as they should, and figure out exactly how to develop drugs to counter them.”
Support for this study was provided by Slim Initiative in Genomic Medicine for the Americas (SIGMA), a joint U.S.-Mexico project funded by the Carlos Slim Foundation.
Paper(s) cited
Dvela-Levitt M, et al. Small molecule targets TMED9 and promotes lysosomal degradation to reverse proteinopathy. Cell. Online July 25, 2019. DOI: 10.1016/j.cell.2019.07.002.